In late October we met at UCLH for a day dedicated to ILD. Thank you to Dr Joanna Porter for putting on such an engaging and relevant programme, and for inviting excellent external speakers. And well done to Alex Mackay for live tweeting the day.

Liver failure
We started the day in slightly unfamiliar territory, below the diaphragm. Prof Julia Wendon from King’s took us on a tour of acute, subacute and chronic liver failure , for an excellent GIM session.

Just in case you forgot (from http://www.liver.co.uk)
Causes of liver failure include:
- primary liver failure – eg paracetamol OD, acute viral hepatitis, ischaemic hepatitis
- chronic liver failure – cirrhotic. Look for ascites, splenomegaly, patent paraumbilical vein on USS (longstanding portal HTN)
- surgical causes – eg hepatectomy, traumatic, liver Bx, ERCP
- secondary liver failure – sepsis, inflammation, systemic disease, drugs (cholestasis)
Critically ill cirrhotics can be scary to treat as they suffer variceal bleeds and metabolic disarray. Often precipitated by alcoholic hepatitis and sepsis. End organ dysfunction can occur – brain, cardiac/vascular, renal, respiratory. In order to keep patients safe when have delirium/encephalopathy, we were advised to use haloperidol (but watch QTc), and avoid opiates and benzos if possible.
Sepsis has a high mortality in acute on chronic liver failure. Pre-emptive (rather than prophylactic) antibiotics are advisable, particularly as a higher mortality is seen with increasing time to antibiotics. Unfortunately CRP is not necessarily helpful (as produced by the liver), and ‘rotating door’ unwell cirrhotics can get multi-drug resistant organisms. A high index of suspicion is needed. AKI confers an additional mortality risk, but avoid nihilism and treat aggressively with terlipressin and albumin as this leads to better outcome. Terlipressin is essentially noradrenaline that can be used safely on a ward. It maintains perfusion of the kidneys. Another way to enhance renal perfusion is to reduce intra-abdominal pressure by draining tense ascites. Of course a sample should be sent to the lab, and if WCC >250 this is diagnostic of SBP.
- Jalan, Rajiv, et al. “Acute-on chronic liver failure.” Journal of hepatology 57.6 (2012): 1336-1348.
- Levesque, Eric, et al. “Prospective evaluation of the prognostic scores for cirrhotic patients admitted to an intensive care unit.” Journal of hepatology 56.1 (2012): 95-102.
- Arabi, Yaseen M., et al. “Antimicrobial therapeutic determinants of outcomes from septic shock among patients with cirrhosis.” Hepatology 56.6 (2012): 2305-2315.
- Fagundes, Claudia, et al. “A modified acute kidney injury classification for diagnosis and risk stratification of impairment of kidney function in cirrhosis.”Journal of hepatology 59.3 (2013): 474-481.
- European Association For The Study Of The Liver. “EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis.” Journal of Hepatology 53.3 (2010): 397-417.
The management of variceal bleeding has changed dramatically over the years. If there is bleeding, think infection, and therefore give antibiotics fast. Then give terlipressin 2mg then 1mg 6hrly. Then call an endoscopist. If a second endoscopy is needed to control bleeding, then refer for TIPS. Early use of TIPS reduces mortality and treatment failure in selected groups.
We were reminded that a restrictive transfusion policy has better outcomes than a liberal policy, and therefore Hb 8 is appropriate. There is no evidence for the use of coagulation factors in this context, and in fact 70% cirrhotics are prothrombotic despite high INR (if use TEG etc). Important markers of higher risk of bleeding are low fibrinogen and low platelets.
Hepatic encephalopathy can be a problem in these patients so it is important to watch their airway. Cirrhotics generally have a shrunken brain so cerebral oedema does not kill them. In hepatic encephalopathy there is also no evidence for protein restriction, and patients should be NG fed if necessary until more alert.
- Villanueva, Càndid, et al. “Transfusion strategies for acute upper gastrointestinal bleeding.” New England Journal of Medicine 368.1 (2013): 11-21.
- Córdoba, Juan, et al. “Normal protein diet for episodic hepatic encephalopathy: results of a randomized study.” Journal of hepatology 41.1 (2004): 38-43.
Alcoholic hepatitis may require different treatment (ie steroids) so is important to recognise. Various scoring systems are available, including the LILLE score, which is complicated, and the Glasgow score, which is easier at the bedside. Score 9+ for steroids as this group has worse survival. Steroids are beneficial, pentoxyphyline is not. But if bilirubin does not fall then stop, as adverse effects outweigh benefits. The usual regime is 40mg Pred for 1-2 wks then reduce over 2/52 if on ward, or 50mg 6hrly hydrocort if on ICU.Hyponatraemia is an additional poor prognostic sign.
- Cordoba, Juan, et al. “Characteristics, risk factors, and mortality of cirrhotic patients hospitalized for hepatic encephalopathy with and without acute-on-chronic liver failure (ACLF).” Journal of hepatology 60.2 (2014): 275-281.
- Stewart, Stephen, et al. “A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis.” Journal of hepatology 47.2 (2007): 277-283.
- Louvet, Alexandre, et al. “The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids.” Hepatology 45.6 (2007): 1348-1354.
- Forrest, Ewan H., et al. “The Glasgow alcoholic hepatitis score identifies patients who may benefit from corticosteroids.” Gut 56.12 (2007): 1743-1746.
Albumin is good in SBP, but has no evidence of benefit in cirrhotics with sepsis other than SBP.
- Sort, Pau, et al. “Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis.” New England Journal of Medicine 341.6 (1999): 403-409.
- Thévenot, Thierry, et al. “Effect of albumin in cirrhotic patients with infection other than spontaneous bacterial peritonitis. a randomized trial.” Journal of hepatology 62.4 (2015): 822-830.
We moved on to consider associations of causes of liver failure with lung pathology. We briefly considered portopulmonary hypertension (painful for those who recently took the SCE, in which both hepatopulmonary and portopulmonary hypertension featured). We were advised that treatment is with the same agents as those used for pulmonary hypertension. If pressure remains high despite treatment then patients have poor outcomes and transplant should be considered.
Patients with hepatopulmonary syndrome are dyspnoeic, clubbed and characteristically desaturate when they sit up (orthodeoxia). This is due to VQ mismatch, and shunting. A contrast/bubble ECHO will reveal the diagnosis. Elevated levels of endothelin-1 are found in hepatic venous blood. Hypoxia in patients with hepatopulmonary syndrome is related to the diameter of peripheral pulmonary vessels. 10% cirrhotic patients will have shunting.
- Hoeper, Marius M., Michael J. Krowka, and Christian P. Strassburg. “Portopulmonary hypertension and hepatopulmonary syndrome.” The Lancet363.9419 (2004): 1461-1468.
- Berthelot, P., et al. “Arterial changes in the lungs in cirrhosis of the liver—lung spider nevi.” New England Journal of Medicine 274.6 (1966): 291-298.
- Koch, David G., et al. “Elevated levels of endothelin-1 in hepatic venous blood are associated with intrapulmonary vasodilatation in humans.” Digestive diseases and sciences 57.2 (2012): 516-523.
- Jalan, Rajiv, et al. “Acute-on chronic liver failure.” Journal of hepatology 57.6 (2012): 1336-1348.
We were advised “Don’t always live within the literature.” Over time survival of cirrhotic patients has improved. This would not have happened if aggressive treatments hadn’t been tied in very sick patients. Patients should have a treatment trial of aggressive treatment and see response. e.g. use ECMO. Aetiology does not make a difference to treatment response.
Extra snippets of liver-related knowledge were:
- Beware, subacute liver failure can look very like cirrhosis on CT. It is therefore important to have the relevant clinical information. If AST >300 think subacute liver failure.
- Hypoxic hepatitis
- Fuhrmann, Valentin, et al. “Hypoxic hepatitis: underlying conditions and risk factors for mortality in critically ill patients.” Intensive care medicine 35.8 (2009): 1397-1405.
- Henrion, Jean, et al. “Hypoxic hepatitis: clinical and hemodynamic study in 142 consecutive cases.” Medicine 82.6 (2003): 392-406.
- Ecstacy is bad news! Diarrhoea = gut failure.
- Kings acute liver transplant referral – pick up the phone.
- NAC has an evidence base in paracetamol – but for 5 days only. It is a potent immunosuppressor (NFKb). EtOH grade 1-2 has evidence. For hypoxic hepatitis there is little evidence. Theoretically it repletes suphydrals and therefore has a possible role.
Welcome
After a great start to the day, we were formally welcomed by Dr Joanna Porter who provided an overview of interstitial lung diseases (ILDs).
ILD is increasing and is also increasingly recognised. There are 30,000+ new cases each year in the UK with 10,000 deaths a year. The BTS has recently started an IPF registry in recognition of the fact that numbers are largely unknown. There is geographical variation with more IPF in the north, but the cause of this variation is unclear. At UCLH 50% are associated with autoimmune disease – scleroderma 90%, dermatomyositis 70%, RA 10%.
The single biggest non-inheritable risk factor for RA is smoking. There is current interest in this, and what it might tell us about mechanisms of disease.
IPF has a high mortality and morbidity with variable prognosis. it is therefore difficult to inform and advise patients. Large groups are needed for trials, and it not always easy to recruit. We need to stratify patients into prognostic groups – but how? Drug development is currently limited by disease heterogeneity and problems assessing response to treatment. The Breathing Matters charity was set up to support patients, and also raise money for research to try to change this situation.
The UCLP ILD service is one of 12 nationally commissioned centres in the UK (4 in London: Brompton, Imperial, GKT, UCLH). At UCLH there is a monthly ILD MDT (2nd Tues of the month), which Dr Porter encouraged us to attend (although not all at once..). It is the fastest growing ILD service in UK. A major benefit of being seen in a specialist service is access to (expensive) drugs which are not widely available. Unique features of the UCLH service include: cryobiopsy (bronchoscopic, relatively non-invasive), satellite transplant clinic Hub, joint Rheumatology clinic (Prof Isenberg), sarcoid MDT, and PET scanning (Prof Groves).
To give some context to the rest of the day we were reminded of the basics of ILDs. We revised what TLCO measured – a look at the interstitial membrane – and what constitutes the alveolar-capillary unit. We had a peek at the histological findings in ILDs, in particular the features of end stage fibrosis. It was heartening to hear that Dr Porter thought that classification of ILDs was a nightmare, particularly since it has changed multiple times over the years. She simplified things by focusing on the inflammatory (leucocytes), fibrotic (fibroblasts) spectrum, and placing various ILDs along this spectrum. This also provided a way into thinking about therapies, which might be anti-inflammatory or anti-fibrotic.
- Organising pneumonia – granulation tissue in alveolar space + interstitial inflammation
- NSIP – ground glass. More fibrotic form. Fibroblasts + inflammatory cells. Blue dotty things = inflammation.
- IPF – fibrotic. UIP. Fibroblastic foci.
Remember that this is a spectrum and different elements can co-exist.
Autoimmune diseases ——–Drugs/dusts —————–Granulomatous —————— Idiopathic
COP —— ————————————Idiopathic NSIP (cellular/fibrotic) —————————————— IPF
We were treated to a story of acute alveolitis due to goose down duvet, apparently common in UCL students in Michaelmas term! Always take a thorough history…..
When considering management, we were encouraged to find the cause and remove it. This is the most successful strategy when possible. After this (or when not possible) the next most important question is whether the process is predominantly inflammatory or fibrotic. Bloods and imaging can help. The next question ishow quickly are things getting worse? For this serial imaging and lung function are required. Then the question becomes what are the treatment options and what is right for this patient?
We finished on a positive note, enthused by Dr Porter’s optimism and hope for a period of enlightenment in ILD!
Radiology of ILD
Next, Dr Tom Semple helped us to appreciate the essential role of the Radiologist in the diagnosis and management of ILD. Specific roles of a specialist ILD Radiologist include:
- confirm pathology
- look for features that suggest specific pathology (both pulmonary and non-pulmonary)
- determine extent of disease and location of disease (?guide biopsy)
- track follow-up.
Extrapulmonary features might include RA visible in the bones, enlarged LA ?amiodarone lung, bright liver ?amiodarone, hiatus hernia/oesphagus ? systemic sclerosis, fibrosing mediastinitis, lymphangitis.
We had a brief physics lesson, and were reminded how CT works and what a Hounsfield unit is. We thought about the difference between modern spiral/ helical scanners, and how single slices were acquired in the old style of HRCT, requiring a higher radiation dose. We marvelled at the vast range of grayscales available, and were saddened that the human eye is incapable of distinguishing many of them, which is why ‘windowing’ on PACS systems is so useful. We refreshed our anatomical knowledge, focusing on the secondary pulmonary lobule, and it’s relevane to determining the causes of ILDs. The secondary pulmonary lobule includes the terminal bronchiole, interlobular septa, and central bronchovascular structure. Centrilobular nodularity therefore indicates an airway centred process such as hypersensitivity pneumonitis. Randomly distributed nodules might indicate miliary TB. Septal thickening and a pleural effusion suggests heart failure. Lobular septal thickening, and beaded/nodular thickening suggests sarcoidosis or lymphangitis. The infamous tree-in-bud indicates that the airway and alveoli are filled in, usually by infection e.g. TB. Distorted parenchyma, and bronchocentric fibrosis, with calcified nodes should make you think of sarcoid. A peaked diaphragm indicates volume loss.
When looking at a CT scan in the context of possible fibrotic lung disease, ask yourself 4 questions:
1. normal/abnormal?
2. is abnormality an interstitial process?
3. is it fibrotic? look for features such as reticulation, traction bronchiectasis, crowded bronchi
4. are there any specific features to suggest an underlying cause?
Dr Temple reminded us that the oblique fissures should come up to the aortic arch; if lower this indicates volume loss. The differential would then be UIP (IPF, RA, other), NSIP, chronic HP, (look for bronchocentricity, as clue) fibrotic organising pneumonia (perilobular sign, reverse halo/Atol sign, sub-pleural dense consolidation). It can be difficult to tell if there is co-existent emphysema or if there are UIP cysts.
We were given an insight into what’s new in CT, including dual source CT perfusion imaging and a lung probe, which is a weird thing that fluoresces via bronchoscopy. V cool!
Whom to Refer and When
Helen Garthwaite (clinical research fellow) provided an in depth insight into the UCLH ILD service. Prior to 2013 individual trusts responded to local PCT requirements. Since NHS restructuring, CCGs have commissioned services at local level (see the Leadership and Management page for links to NHS structure diagrams). ILD is outside this structure as it is a specialist nationally funded service. Features of a specialist service are: relatively few hospitals provide a service with specific expertise; services are accessed by comparatively small numbers of patients; the catchment is >1million. There would be financial implications for diagnostics and treatments for CCGs if specialist services were provided locally. In Respiratory, specialist commissioned services include ILD, difficult asthma and home NIV.
The aims of a specialist ILD service (see NICE) are:
- allows equality of access to a specialist MDT diagnosis (improves diagnostic accuracy)
- equal access to current treatment modalities and disease specific management plans
- oversee aspects of care that may be a challenge for local units e.g. administration of cytotoxics
- identify individuals requiring referral to lung transplant service – discuss 3-6/12 after diagnosis, and should be seen within 1 month
- integrate with other services e.g. palliative care
- reduce morbidity and mortality, including reducing hospitalisation
An ILD MDT consists of a respiratory physician, a radiologist, a histopathologist, an ILD spec nurse, and a surgeon where appropriate.
Clinical trials are also key to the UCLH service. Several trials are currently ongoing or recruiting at UCLH. HRCT is integral to the diagnosis of ILD; it gives excellent structural information but is static and is unable to provide any comment on function. It is taken at a single point in time, and not reliably prognostic. Lung function tests are also not necessarily helpful in prognosis. IPF (like many Respiratory conditions) has a relatively unpredictable prognosis. There are probably at least 3 phenotypes, and 20% live >5yrs. PET HRCT is acquired at baseline and post-treatment (if applicable). Patients must be over 40yrs and be able to comply with the PET HRCT protocol. There is increased FDG avidity observed in the lungs with ILD (NSIP/IPF), with prognostic implications. This is interesting as it suggests an active process (rather than ‘burned out’ fibrosis) and therefore gives hope for interventions. Future research is to include a systemic sclerosis cohort pre-post nintedanib, with the aim of determining the level of minimal important difference, and to gain insights into the source of the signal.
The coagulation cascade is an early initial response to injury (fibrin, thrombin), and an area of interest in ILD research. The cellular effects of thrombin are via PAR binding, releasing fibrogenic cytokines – TGFB, CTGF, PDGF-AA. IPF Warfarin trials were negative with excess deaths, but these were patients without VTE. It is hoped that newer anticoagulants could have a direct/specific effect.
TIPAC1 was an RCT of septrin 960mg BD vs placebo in IPF. It showed no difference in FVC, DLCO, 6MWT, or MRC score. However, on a per-protocol analysis there was a reduction in percentage requiring oxygen and in all-cause mortality. The use of septrin reduced respiratory tract infections (? a trigger for decline) but increased the incidence of nausea and rash. TIPAC2 aims to reproduce this finding in 330 IPF patients over 30 sites (of which UCL is one site). Inclusion criteria include a definite diagnosis of IPF (biopsy not necessarily required if radiology typical), FVC <70%, no immunosuppression (up to 10mg Pred).
In answer to the question who to refer to the ILD service and when, we were encouraged to refer anyone with any doubt about diagnosis, and anyone for trials or treatments not accessible locally. Members of the ILD service are very happy to discuss cases prior to a formal referral if there is any doubt.
Transbronchial Cryogenic Lung Biopsies in the diagnosis of ILD
Next up was Dr Theresia Mikolasch, with a much-anticipated talk on cryo lung biopsies. We began by considering VATS biopsy, the traditional route to definite diagnosis, and its’ associated problems.
A 4.8% mortality rate and 14.8% mean complication rate are high. It is possible to risk stratify but the risks remain significant and some aspects are unpredictable. In 2012 910 patients underwent surgical lung biopsy in the UK, spending 1.5 patient-years in hospital (data from yearly audit from RCSEng). Data is also available from a single centre (in Scotland) including 103 patients. 4.9% died within 30 days, and no definitive pathological diagnosis was reached in 30%.
So, does getting a tissue diagnosis matter? Yes it does! There are now 2 (almost) drugs available to treat IPF: Pirfenidone and Nintedanib. Unfortunately, using radiology for diagnosis has limits as the end stage of many processes looks the same. However, different treatments may be beneficial, even when the radiological appearances are fibrotic.
So, what is cryo lung biopsy? NO is pumped through a probe and freezes tissue.
Traditionally forceps biopsy was used, but this is no good as tissue architecture is ruined (crush artefact, haemorrhage artefact, small biopsy area). In a cryobiopsy the lung architecture is perfectly preserved, and there is a greater area of biopsy, facilitating accurate diagnosis.
There are of course complications and risks. These include pneumothorax, moderate/major bleeding (this technique is contraindicated if the patient has pulmonary hypertension, or is on anticoagulation, and patients are pre-medicated with iv tranexamic acid with adrenaline used endobronchially). Pneumothorax rate in published literature is variable, but up to 20% in fibrotic patients. The potential advantages of cryobiopsy over surgical biopsy are:
- day case procedure for most
- lower mortality
- lower exacerbation rates
- do not have to be ‘surgical candidate’
- no scars or long term neuropathic pain
- lower cost
The LUNG COOL Trial (CryOextraction of Lung tissue for diagnosis if ILD) includes patients who are referred for VATS lung biopsy. They have a cryobiosy first and are then discussed in MDT. This trial is aimed at determining cost-effectiveness of cryobiopsy. There is also a direct comparison trial of VATS vs cryobiopsy planned. Recruitment is challenging as there are only small numbers of VATS referrals.
Following this fab talk we had a brief discussion on other complementary technologies. We considered how alveoscopy (ultrafine confical microscopy on the end of the probe) could be used to target biopsies. We also talked about the value of xray screening in checking the location of the cryobiopsy.
If you want to know more about cryobiopsy why not watch this great video: www.erbe-med.com

From: http://www.erbe-med.com
If you want to watch this ‘cool procedure’ in real life, contact Theresia to arrange a date.
PET scanning: a HOT topic in ILD
Prof Ashley Groves convinced us all of the value of a nuclear medicine department! At the Institute of Nuclear Medicine at UCLH, over 20,000 clinical examinations are completed a year! The first PET-MR in the UK is owned by the department, and they have some other fancy kit too. FDG-PET is the most commonly performed examination in respiratory patients. It is an exquisitely sensitive (pico molar) molecular imaging technique. FDG = 18 radio-labelled glucose. Unfortunately, there are a lack of biomarkers in IPF. Could molecular imaging help?
Surprisingly there is high signal in areas of fibrosis – it is highly metabolically active. This gives us hope that treatments and disease modification is possible. Metabolic activity is quantified by region of interest analysis. There is a significant correlation between pulmonary FDG uptake and FVC and TLCO % predicted at diagnosis. There is also correlation with SGRQ general activity score. Thus far this is mainly a research tool, but the UCLH team are developing experience and expertise in using PET-CT in ILD to facilitate clinical decision making. Short term reproducibility has been confirmed for FDG- PET in ILD. It is as yet unclear what happens to patients longterm who are NOT treated. Dr Porter discussed how she finds FDG-PET particularly useful when there are difficult treatment decisions. Patients with fibrotic NSIP may be treated with cyclophasphomide. A repeat PET-CT may provide additional information to determine whether to continue this or not, or switch to pirfenidone. Helpful for patients as provides additional data to gauge success when committing to a treatment. How does it compare to 6MWT? This is as yet unknown.
There are other forms of molecular imaging including PET-MR (in the Macmillan centre). There is the potential to use different types of MR sequence e.g. diffusion imaging. This technique has a reduced radiation dose. 68GA DOTATATE is an alternative ligand (somatostatin-2). Activated macrophages over-express somatostatin receptors so this may be of interest. Future developments may include using different ligands eg integrin tracer to measure fibroblast metabolism.
CTD-associated ILD
Dr Maria Leandro took us on a tour of CTD-associated ILDs. In systemic sclerosis (SScl) it occurs in 40%. In poly/myositis (PM/DM) it occurs in 30-70%, in RA 10-58%. ILD can be the first or only manifestation of a CTD.
It is important, as it provides information on the natural history, helps the clinician determine the need/frequency of monitoring, the likely response to treatment, and prognosis. It can be challenging to identify a CTD-associated ILD with confidence. An MDT which includes a Rheumatologist can be helpful.
The main question is usually “Is this ILD associated with systemic autoimmunity?” In most patients a detailed clinical history and examination, then selected autoantibodies and muscle enzymes are helpful and sufficient. Joint clinics/MDT discussion including a Rheumatologist can enhance this further. Extra-thoracic manifestations can be a clue: Raynaud, swollen fingers, oesophageal dysmotility, sicca syndrome, arthralgia, hyperkeratosis of hands/fingers.
Recommended investigations:
- ANA (titre and immunofluoresence pattern) -> anti-centromere, anti-dsDNA
- ENA: anti-Ro, -La, -Sm, -RNP, -Scl70, -PM/Scl70, -Jo1
- Other SScl assoc e.g. anti-U3-RNP, anti-RNA, polymerases I, II anti-Th/To
- Myositis specific – anti-Jo1, -PL12, PL7, -Mi2….pts ANA negative.
- RF, anti-CCP
- HRCT: NSIP is more suggestive of CTD-ILD (but others seen)
- Histopathological features: dense perivascular collagen deposition, lymphoid aggregates with GC formation
With more thorough evaluation/follow-up 15% of those with ILD had associated CTD.
- Mittoo, Shikha, et al. “Ascertainment of collagen vascular disease in patients presenting with interstitial lung disease.” Respiratory medicine 103.8 (2009): 1152-1158.
- Strange, Charlie, and Kristin B. Highland. “Interstitial lung disease in the patient who has connective tissue disease.” Clinics in chest medicine 25.3 (2004): 549-559.
- Watanabe, Kizuku, et al. “Detection of antisynthetase syndrome in patients with idiopathic interstitial pneumonias.” Respiratory medicine 105.8 (2011): 1238-1247.
In whom is it worthwhile testing for an extended antibody panel?
- Women <50yrs, anyone with extra thoracic manifestations that are suggestive.
- NSIP, LIP, or other suggestive pattern
- ANA or RF if high titre, or nucleolar-staining ANA
- anyone with other suggestive antibodies e.g. RNP
We were introduced to the concept of lung-dominant CTD, and the difficulties of definitions.
- Fischer, Aryeh, et al. “Connective tissue disease-associated interstitial lung disease: a call for clarification.” CHEST Journal 138.2 (2010): 251-256.
- Pereira, Daniel Antunes Silva, et al. “Lung-dominant connective tissue disease among patients with interstitial lung disease: prevalence, functional stability, and common extrathoracic features.” Jornal Brasileiro de Pneumologia 41.2 (2015): 151-160.
Undifferentiated CTD is usually considered mild by Rheumatologists, but they may underestimate the impact of the ILD. Some criteria have been suggested and a classification system:
UCTD ————–> autoimmune ILD —————-> LD-CTD
Nailfold capilaroscopy can be helpful in the right hands – when scleroderma type changes can be identified. MDT collaboration becomes even more important in caring for patients with overlap syndromes, and in those in whom a treatment controls 1 aspect of disease (e.g. joints) but not other aspects (e.g. falling TLCO).
Is answer to a question about the benefit of repeating autoantibodies, Dr Leandro advised that there was only benefit if something significant had changed, particularly as patients with 1 autoimmune disease can develop another one. It is best to keep an open mind if you have not made a diagnosis and there is progression when the condition is thought to be idiopathic e.g. COP.
Current Pharmacological/non-pharmacological therapies for NSIP and IPF
Dr Joanna Porter and Dr Helen Booth gave us more insight into the UCLH ILD service. It treats 1000 patient events/yr. In terms of ILD 17.5% idiopathic, 37.5% sarcoidosis, 45% identified aetiology. Of UIP-type radiology 46.4% idiopathic – IPF. 12% sarcoidosis. 41.6% identified aetiology. THis is perhaps an unusual spread due to the particular interest in CTDs by the Rheumatologists at UCLH, and the historic interest in sarcoid by Prof Spiro, and now Dr Booth.
We went back to basics and considered which immunopathologies go the lung.
- innate immune defence – neutrophils, MMPs
- adaptive immune response – type I = IgE allergic asthma, type II – Goodpasture’s syndrome, type III = immune complex mediated, type IV = TB, sarcoid.
Type II hypersensitivity reactions include Goodpasture’s syndrome. Autoantibodies against type IV collagen lead to renal disease and lung disease, through antibody-mediated cell toxicity. This is probably what happens in RA – 30% have lung disease, which correlates with ACPA (anti-citrullinated antibodies).
Type III hypersensitivity reactions are mediated by immune complexes. In Hypersensitivity Pneumonitis contact with birds, farmers, occupational hazards leads to disease. African grey parrots seem to be particularly popular in Bloomsbury…. In HP an abnormal BAL may be seen- lymphocytosis, high CD8:CD4 ratio. Circulating precipitin antibodies may identify the cause as HP but do not correlate with severity. Pathologically there is B cell activation, T cell infiltrate and granuloma-like clusters in the interstitium. Multinucleate giant cells are also seen. We considered whether HP could be a cause of many cases of idiopathic ILD? Difficult to answer.
Type IV delayed hypersensitivity occurs in TB, fungi, parasites, and transplant rejection.
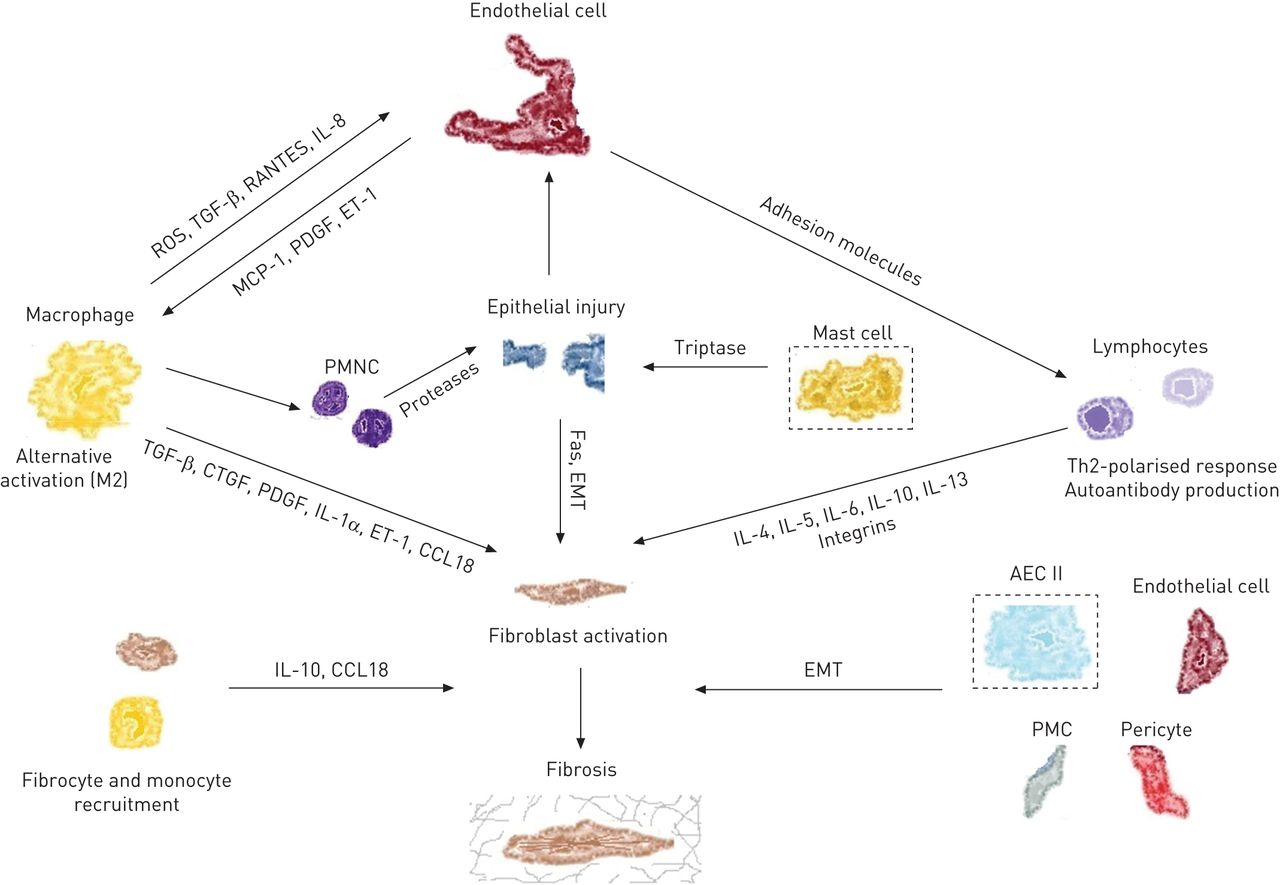
The aetiology of IPF remains unclear. There is alveolar-capillary unit injury but what is the primary event? Endothelial damage? Alveolar cell damage? Injury is followed by fibroblast influx and associated with neutrophil activation. There are many potential molecular targets including TGF-B, CXCL12, MMP, endothelia, and the myofibroblast. It is unclear exactly how pirfenidone woeks, but it may act through blocking action of TGF-B. Nintedanib is a tyrosine kinase inhibitor.
We were reminded that investigations in ILDs are targeted at finding the causes that can be modified. It would be nice to do a BAL in all patients (200-240ml in 4 aliquots, and should get back 30% return) to look for significant lymphocytosis 30% – suggests a cause other than IPF. CD4/CD8 ratio should be up in sarcoid and down in NSIP – true?
We moved on to consider management in NSIP:
- Supportive care including pulmonary rehab, stop smoking, oxygen.
- Drugs
- steroids – high doses justified in NSIP/HP/OP, less so in fibrotic NSIP
- cyclophosphamide – monthly iv. give 3 monthly doses and reassess. Reluctant to give in elderly
- Azathioprine – prodrug for mercaptopurine (TPMT levels) – perhaps slower
- MMF – antilymphocyte and anti-fibroblast proliferation
- cyclosporine – inhibits calcineurin in T cells
- MTX – DHFR clock and other effects
- colchicine?
- new biologics – antiTNF, rituximab (in development/trials)
- pirfenidone, nintedanib – not for NSIP at present. Future?
- Don’t treat everyone – consider entry to clinical trials. Consider for lung transplant.
- Palliative care – alongside active treatment. Should include advance directives and ventilation discussion.
For IPF some of the principles are the same
- Supportive care, including pulmonary rehab (Cochrane), and oxygen.
Level of dyspnoea often less than COPD. Often need higher flow rates than COPD. - Stop smoking. In studies of IPF – ASCEND, INPULSIS, TIPAC there are relatively high rates of smokers pre-diagnosis, but low rates of ongoing smoking. This is in contrast to COPD in which there is lots of continuing smoking (eg Wisdom).
- Flu vaccine is very important. Under 65 at risk groups do not have great uptake. Something to work on.
- Drugs:
- Warfarin in IPF trial was stopped prematurely at 32wks due to excess deaths in the warfarin group.
- Pirfenidone – CAPACITY 1 and 2: a change in FVC was seen in CAPACITY 1 but not 2. EU approved 2011, NICE approved 2013. FDA approved Oct 2014 after ASCEND achieved endpoint of change in FVC. NICE approved Perfenidone for IPF with FVC 50-80%. It should be discontinued if there is disease progression (decline >10% in 12 months). SE nausea, rash. Side effects must be proactively managed – take drug with food, wear sunscreen and cover skin in sun.
- Noble, Paul W., et al. “Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials.” The Lancet 377.9779 (2011): 1760-1769.
- King Jr, Talmadge E., et al. “A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis.” New England Journal of Medicine 370.22 (2014): 2083-2092.
- Martinez, Fernando J., et al. “Randomized Trial of N-acetylcysteine in Idiopathic Pulmonary Fibrosis.” The New England journal of medicine 370.22 (2014): 2093.
- Nintedanib – TOMORROW and INPULSIS 1 and 2 – change in FVC, reduction in mortality. FDA approved Oct 2014. EU approved Jan 2015. Anticipated NICE publication 2016.
In the future combined treatments may be used. Studies on pharmacokinetics are ongoing at UCLH.
In the TIPAC study of cotrimoxazole there was an all cause mortality reduction in per-protocol analysis (not intention to treat). There were also lots of adverse events – GI or skin. TIPAC 2 is now open – UCL is a recruiting centre.
We then segwayed into a discussion about Vitamin D in which Dr Booth reminded us of the feedback lops and highlighted the fact that we should be measuring 1,25-dihydroxycholecalciferol but this cannot be tested in most labs.
ILD presenting as severe respiratory failure
Dr Boris Lams (Guys and St Thomas’) gave us an impressive insight into the treatment of the most severe forms of ARDS, in which he made putting patients onto ECMO sound as easy as putting in a cannula! Maybe it is for him…
He focused on the difficulties of diagnosing ILD presenting as respiratory failure, and therefore getting the right treatment early. Many patients referred for ECMO are initially thought to have pneumonia and ARDS. Some patients with ARDS do have a relatively clear cause e.g. pancreatitis, meningococcal sepsis. Other’s don’t.

Not everything labelled as pneumonia and ARDS is this. Clues include: haemoptysis, a prodrome of arthralgia, relative sparing of some areas on CT, lack of multi-organ dysfunction syndrome (i.e. no inotropes, not in AKI)
An example case had ANCA positive GPA. The patient was retrieved on ECMO, when they had diffuse alveolar haemorrhage. They were given Prednisolone and cyclophosphamide as well as ECMO. Previously such patients did badly due to ventilator-associcated damage. This patient was off ECMO after 10/7 and was much better!
If pneumonia doesn’t get better and the patient has haemoptysis, think PVL-Staph or ANCA-associated vasculitis.
The practice is now to give iv methylprednisolone, then plasma exchange, then Rituximab, then wait, then do more plasma exchange, then more Rituximab. This regimes has had great outcomes in ANCA-associated vasculitis.
We were reminded that a raised CRP does NOT equate to infection.
Imaging clues to alveolar haemorrhage are that it spares the edges on CT, and is fluffy/cloudy.
Dr Lams shared some difficult cases, including a patient with antibody negative GBM disease! The team had to stop plasma exchange and give IVIG. This patient had a great outcome. He described a case of acute eosinophilic pneumonia, presenting with fever, raised WCC and CRP and rapid progression to respiratory failure. There is usually a history of some smoking. Patients may not have high eosinophils at presentation but develop them. Treatment involves iv methylprednisolone. If pulsed ivMP is not effective ECMO can be an option with good outcomes!
The last case was a patient with anti-Jo1 and SSA +ve, with normal CK. Clinically this was a myopathic dermatomyositis. Clues included single organ failure (therefore not pneumonia and sepsis), ragged cuticles, and a prodrome of myalgia.
Case selection for ECMO is key. In a case series those who had a highly inflammatory picture and a short history pre-ECMO did better. Advice from Dr Lams was to reconsider the diagnosis (ie pneumonia) when it doesn’t progress as expected, to actively look for those who will respond to immunosuppression, and give it early. And for those that need it, to get them onto ECMO quickly. And to have a constant state of low grade anxiety!
Don’t forget Poiseuille (the airway)
Dr Jeremy George finished the day with an entertaining and informative look at upper airway involvement in ILDs. This is rare, and often missed or misdiagnosed. Often upper airway involvement is misdiagnosed as asthma. This can potentially have devastating clinical consequences and result in litigation. A key message was always to reconsider the diagnosis if things are not going as expected.
Central airway strictures may occur in some ILDs – GPA 25% Sarcoidosis 15%.
Dr George passed on some tips on how to avoid missing central airway obstruction:
- have a high index of suspicion
- consider in any patient unresponsive to asthma treatment
- listen for a monophonic wheeze or stridor
- look for a disproportionately low PEF in relation to the FEV1
The Empey index is extremely useful: FEV1 (ml/sec) / PEFR (L/min). <10 normal >10 abnormal
15% pts with GPA get localised endobronchial disease. Subglottic stenosis is most common. There is probably a higher proportion but some with milder symptoms are likely to be missed/assumed to be asthma.
We heard the story of a 14yr old girl who had a week of flu-like symptoms and then developed bilateral infiltrates. She was diagnosed with pneumonia but had a poor response to antibiotics and was intubated and ventilated. c-ANCA rose and the patient was thought to have GPA and was treated with plasma exchange, ivMP and cyclophosphamide. She got better and was discharged after 1/12. The patient was followed-up by a paediatric nephrologist who weaned off the immunosuppression. She also underwent repeated lung function tests, and was followed up by a chest physician. At age 18 she had chest tightness and breathlessness. A salbutamol inhaler was prescribed by the GP. PEFR age 14 was 500L/min, but at age 18 PEFR was 200L/min. The flow-volume loop was also abnormal when reviewed retrospectively. The patient was treated for asthma and the upper airway obstruction was missed until the patient was pregnant and had pre-eclampsia. She was reviewed by an anaesthetist who made the diagnosis of upper airway obstruction. Unfortunately by this time she had a mature scar tissue stricture and was therefore not appropriate for bronchoscopic treatment and required reconstructive surgery. She was awarded £600,000 damages. A cautionary tale…
Upper airway strictures were traditionally treated with immunosuppression, then surgery/tracheostomy.
Modern bronchoscopic approaches include intralesional steroids with balloon dilatation. As a result the requirement for tracheostomy has reduced from 40-50% to 0%. However, it is essential to intervene before mature scar formation. Subglottic strictures can undergo CO2 laser with good effect. For endobronchial strictures it is not possible to use CO2 laser. Instead the stricture is injected with ivMP and then dilated with balloon-cutting balloon bronchoplasty.
Poiseuille’s law states that flow is proportional to the fourth power of diameter, therefore a small improvement in diameter makes a big difference to flow, and therefore to symptoms.
The chance for post-training day drinks and socialising was very much appreciated! See you at the next one.

Discussion
No comments yet.