In November The Royal Free Hospital hosted an innovative programme, themed around Respiratory and Rheumatological overlap conditions. Many thanks to Dr James Goldring, Dr Richard Stratton and everyone at the Royal Free for co-ordinating and contributing to the day. Many thanks to Dr Catherine Rang for sharing notes from the day.

Interstitial lung disease classification and management
Dr Elizabeth Renzoni, started the day with an update on ILD, which is classified into three broad groups:
- Idiopathic
- Associated with systemic involvement
- Associated with a cause/exposure
Within the group associated with a cause/exposure (HP) birds, then moulds are the most common causes in the UK. The ‘idiopathic’ group contains a number of distinct entities and is further sub-classified in the 2013 Revised ATS/ERS Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias:

Although classically associated with smoking, up to 1/3 of patients with DIP are (current?) non-smokers. IPF in the UK has a 20% overall 5yr survival. Latest BLF figures estimate that in 2012, about 32,500 people had IPF in the UK. In 2012, 5,292 people in the UK died from pulmonary fibrosis (0.9% of all deaths and 4.6% of deaths from lung disease), up from 3,964 in 2008.
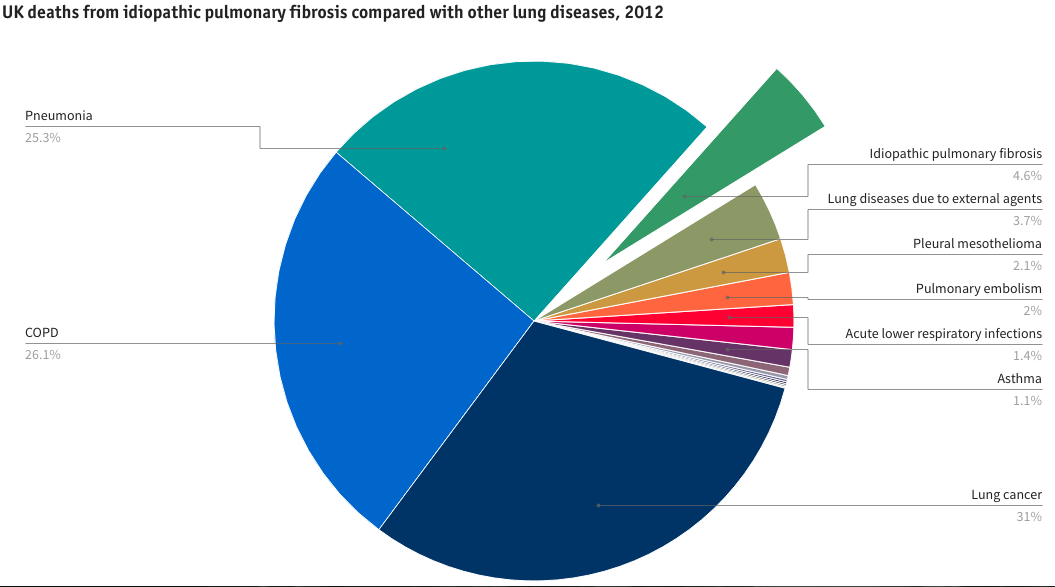
From BLF, Lung Disease in the UK. http://www.statistics.blf.org.uk/pulmonary-fibrosis
3-20% of IPF is familial and is commonest in Caucasians. A single-nucleotide polymorphism (SNP) (rs35705950) in the promoter of the gene encoding mucin 5B (MUC5B) has been shown to be associated with both familial interstitial pneumonia and sporadic idiopathic pulmonary fibrosis. Read more about promoter MUC5B:
- Hunninghake, Gary M., et al. “MUC5B promoter polymorphism and interstitial lung abnormalities.” New England Journal of Medicine 368.23 (2013): 2192-2200.
Fibrotic ILDs:
- ¼ IPF (only ~ 60% have definite UIP on CT imaging)
- ¼ CTD-ILD
- ¼ Sarcoid
- 10-20% have unclassifiable ILD
The use of transbronchial biopsies in the diagnostic process for ILD: ~83% positive biopsies.
Subacute/ Acute IP: OP/DAD
- seen in idiopathic inflammatory myositis, RA, lupus, Sjogrens
- need to exclude infection
- consider drug reactions (especially in RA)
- early vigorous treatment
Rituximab can be used as rescue therapy: anti-CD20 – tends to stabilise previously progressive lung disease. Retrospective series suggest this may be an effective strategy. An prospective RCT is ongoing:
- Sharp, Charles, et al. “Rituximab in autoimmune connective tissue disease–associated interstitial lung disease.” Rheumatology (2016): kew195.
- Keir, Gregory J., et al. “Severe interstitial lung disease in connective tissue disease: rituximab as rescue therapy.” European Respiratory Journal 40.3 (2012): 641-648.
- Rituximab Versus Cyclophosphamide in Connective Tissue Disease-ILD (RECITAL) – RCT comparing Rituximab 1g given on 2 occasions vs monthly Cyclophosphamide for 6/12.
RA + classical UIP can have a similar progression to IPF, whereas other CTD-ILD has a better prognosis.
Pirfenidone for IPF – has shown a borderline improvement in survival. However, there is insufficient survival data and it appears to act more in reducing the rate of decline in lung function (it halves FVC decline).
- Azuma, Arata, et al. “Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis.” American journal of respiratory and critical care medicine 171.9 (2005): 1040-1047.
- Taniguchi, H., et al. “Pirfenidone in idiopathic pulmonary fibrosis.” European Respiratory Journal 35.4 (2010): 821-829.
- Noble, Paul W., et al. “Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials.” The Lancet 377.9779 (2011): 1760-1769.
Use of radiology in interstitial lung disease
Dr Joseph Jacob reminded us f the central place of Radiologists in the diagnosis of ILDs. The positive predictive value of CT in the diagnosis of UIP is high (70-100%). When describing imaging features, the term ‘UIP pattern’ is often used, which has specific diagnostic criteria on HRCT from 2011 (agreed by ERS, ATS and others). HRCTs are classified into: UIP pattern (definite), possible UIP pattern, or inconsistent UIP pattern.
For ‘UIP-pattern’ to be ascribed, the 3 definite features must be present, and none of the ‘inconsistent with UIP’ features (see Radiopedia article for summary).
- subpleural, basal predominence
- reticular abnormality
- honeycombing +/- traction bronchiectasis
Beware of false +ve identification of honeycombing (traction bronchiectasis; emphysema with oedema/infection superimposed; other cystic conditions eg LCH), and also of difficulties with GGO – contrast can cause the imaging to appear as though there is GGO.
Chronic HP is a difficult diagnosis to make on CT imaging, and may be difficult to distinguish from IPF. The changes seen are:
- airtrapping
- nodules
- lobules of decreased attenuation in spared lung (non-fibrotic), leading to mosaicism
- septal thickening
- bronchocentricity in the upper lobes
The evidence of parenchymal vessels is the strongest predictor of mortality.

From Radiopaedia
Opportunistic infections in patients receiving biologic therapy
Dr Marc Lipman reminded us of the importance of recognising that patients receiving biologic therapy are at increased risk of opportunistic infections.
- Novosad, S. A., and K. L. Winthrop. “Beyond tumor necrosis factor inhibition: the expanding pipeline of biologic therapies for inflammatory diseases and their associated infectious sequelae.” Clinical Infectious Diseases (2014): ciu104.
Pneumonia: in patients with RA the rate is 4/100 patient-years vs 2.4 in the general population.
- Winthrop, K. L., et al. “Opportunistic infections and biologic therapies in immune-mediated inflammatory diseases: consensus recommendations for infection reporting during clinical trials and post-marketing surveillance.” Annals of the rheumatic diseases 74.12 (2015): 2107-2116.
TB ~50% is extra-pulmonary if on biologics. TNF alpha blockers: RR for TB is anywhere between 1.6-25
IGRA: T Spot (looks at the specific number of T cells) – indeterminate <2-21%; conversion ~10%. Quantiferon-Gold (3 tubes) moving to Quantiferon Plus (4 tubes) – indeterminate <2-36%; conversion ~10%
- Lake, M. Alexandra, et al. “‘” Why me, why now?” Using clinical immunology and epidemiology to explain who gets nontuberculous mycobacterial infection.” BMC medicine 14.1 (2016): 1.
Management of pulmonary involvement in systemic sclerosis
Prof Christopher Denton updated us on Systemic Sclerosis (SSc). Survival is determined by subset and organ-based manifestations. Currently fibrosis and PH are the major causes of death.
- Diffuse SSc ~ 50% have fibrosis at 15 years; PH equal frequency in both groups
- Limited SSc (which is twice as common as diffuse SSc) ~ 25% of patients have fibrosis
Shortness of breath is major symptom in these patients, which is often multi-factorial – cardiorespiratory and musculoskeletal deconditioning (not to be forgotten) are some of the major causes. Scoring systems are used to assess the extent of the lung disease seen on CT imaging. When lung disease on CT >20%, there is an impact on survival, which deteriorates even further when these changes are >40%. SSc associations with severe lung fibrosis include: diffuse pattern of skin involvement; rapid DTPA clearance; and high KL-6 levels.
Early detection is key. Athol Wells and colleagues have proposed a simple staging system for SSc-ILD as limited or extensive disease, based on simplified HRCT evaluation and FVC estimation. They state that this provides more powerful prognostic information than either component in isolation:
- Goh, Nicole SL, et al. “Interstitial lung disease in systemic sclerosis: a simple staging system.” American journal of respiratory and critical care medicine 177.11 (2008): 1248-1254.
Immunosuppression: iv cyclophosphamide (600mg/m2); oral MMF (2g/day) or azathioprine (150mg/day); prednisolone 10mg od; consider rituximab for rescue therapy
- Tashkin, Donald P., et al. “Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial.” The Lancet Respiratory Medicine 4.9 (2016): 708-719.
This trial compared cyclophosphamide and MMF – statistically these 2 treatments were the same; DLCO improved more quickly in the MMF arm and more deaths were seen in the cyclophosphamide arm. Treatment effect was modest.
Newer treatments are awaited. Autologous haemopoietic stem cell transplantation may improve long term survival, with a modest improvement in lung function. There was a survival benefit after 2 yrs in recent studies (10% mortality initially).
- Volkmann, Elizabeth R., and Donald P. Tashkin. “Treatment of Systemic Sclerosis–related Interstitial Lung Disease: A Review of Existing and Emerging Therapies.” Annals of the American Thoracic Society 13.11 (2016): 2045-2056.
Update on pulmonary hypertension
Dr Benji Schreiber updated us on the classification of pulmonary hypertension:
- PAH
- Idiopathic (mean survival 9 years)
- Familial – BMPR2 mutation – penetrance 20-30%. Promotes an exaggerated inflammatory response and is associated with worse survival and RV function
- Related to CTD (mean survival 4yrs), HIV, portal hypertension, haemolytic anaemia
- Pulmonary veno-occlusive disease: extensive and diffuse occlusion of PV by fibrous tissues. This has a strong association with chemotherapeutic agents, especially cyclophosphamide. These patients are more compromised – have lower saturations and DLCO
- Left heart dysfunction
- Atrial or ventricular
- Valvular
PAWP >15mmHg
- Lung disease
- COPD
- ILD
- Sleep disordered breathing
Do not benefit from vaso-occlusive treatment
- CTEPH
2-4% of PEs will develop into CTEPH. Pulmonary endarterectomy is the main treatment option. Balloon pulmonary angioplasty is increasingly used (mainly used in Japan where they have limited access to CTEPH surgery but this technique has been recently started at Papworth).
- Miscellaneous
~20% of PH patients referred to the service have treatable disease. Echo is a poor tool for identifying PH, and proBNP is a late marker, so if suspected, further investigation is warranted. Vasoreactivity is initially tested with nifedipine.
Current Treatments:
- PDE5 inhibitors : sildenafil, tadalfil (1st line in the UK)
- ERA: bosentan, macitentan
- Soluble guanylate cyclase activator: riociguat (funded in the UK only for inoperable or residual CTEPH)
- Oral IP
CTD associated PAH is different:
- clinical phenotype different:
- -less dramatic haemodynamics
- -higher NTproBNP, lower walk, worse outcome
- opportunity for screening
- lung disease more common
- vasodilator response not seen
- PVOD more common
- Some cases may respond to immunosuppression
Sarcoid diagnostics
Dr James Goldring rounded off the talks with an interesting look at sarcoidosis. The aetiology of sarcoidosis is unknown. It often presents non-specifically and there is a wide spectrum of disease severity. It can follow an unpredictable course and there is no reliable biomarker. Long term follow up is necessary as patients can relapse up to 3 years later. Some patients have treatment refractory disease. These patients can be difficult to manage and there are no standard investigation or treatment guidelines.
- Valeyre, Dominique et al. “Sarcoidosis.” The Lancet, Volume 383, Issue 9923, 1155 – 1167
Increasingly tissue diagnoses is sought, but it can be difficult to obtain biopsy samples as there is often no peripheral tissue. Endobronchial/transbronchial bx (80% sensitivity for stage II/III disease, 10% pneumothorax) by bronchoscopy or EBUS may be required. Laryngeal and tracheobronchial sarcoid needs localised treatment.
EBUS has become increasingly popular, but has limitations in sarcoid diagnosis:
- negative EBUS may need repeating (low NPV in symptomatic patients)
- not all granulomatous LN is sarcoid
- Caseating Granuloma & Non-Caseating Granuloma are not enough to distinguish between sarcoid & TB
- Culture rate for TB LN 49%
- Combined TB-sarcoid is a challenge
- Sarcoid reaction in malignant nodes can occur
- HP – can mimic sarcoid
- Bartonella, brucella, toxoplasmosis, histoplasma – mimic stage II
- IgG4-related disease
- GL-ILD (Granulomatous-Lymphocytic ILD)
Murray, J., et al. “P183 Endobronchial Ultrasound And Tuberculosis: Beware The Non-caseating Granuloma.” Thorax 69.Suppl 2 (2014): A156-A156.
Some feel that EBUS is being overused for this indication, partly driven by the profit to the Trust/department from having an EBUS service. In addition, medicine may be becoming more defensive, with clinicians less willing to make a presumptive diagnosis in the absence of tissue. A biopsy may not be necessary, providing the patient does not have HIV or a previous malignancy. Judgement is required, and further case series and other evidence will aid decision-making in the future.
Definite diagnosis can be made on the basis of:
- Løfgrens syndrome
- Asymptomatic BHL (stage 1) NNB (number needed to biopsy) =1800, especially if lymphadenopathy is symmetrical and non-necrotic or the patient has a history of uveitis –> this is very reasssuring
- Consider not biopsing in stage 2 disease where HRCT is pathognomonic
The day concluded with the London Rheumatology Research Club, including presentations entitled “Role of c-Kit positive cells in pulmonary fibroblast invasion” from Dr Oseme Etomi & Ms Bahja Ahmed-Abdi, UCL, and “B-cell depletion: a 360degree feedback and Rituximab: an appraisal” from Dr Venkat Reddy, UCL.
Thanks again to all involved in organising the day, and for Catherine for the great notes.
See you at the BTS Winter meeting, or at the next Training Day in January (after which there will be drinks, so make sure you get a pass for the evening!).

Discussion
No comments yet.